
前言 急性肾损伤(AKI)是危重症患者的常见并发症,每年导致约200万人死亡,常见诱因包括手术、脓毒症、创伤及肾毒性药物等导致的肾缺血。AKI不仅本身具有致命风险,更是慢性肾脏病(CKD)发展的明确风险因素,然而其背后的病理机制尚待阐明。已知组织缺氧是受损肾脏的显著特征,能够调控实质细胞与免疫细胞的功能;而内皮细胞虽具备重要的免疫调节能力,但在这种失调的氧感知环境中如何应答,目前研究仍然有限。 近日,美国西北大学Pinelopi Kapitsinou团队在《Kidney International》发表了一篇题为《Endothelial prolyl hydroxylase 3 mitigates maladaptive inflammation to promote post-ischemic kidney repair》的研究。该研究表明,内皮细胞中的脯氨酰羟化酶3(PHD3)是缺血后肾脏修复的关键正向调节因子,它通过缺氧诱导因子(HIF)依赖的机制,抑制过度的干扰素-γ(IFN-γ)驱动的内皮炎症反应,进而促进适应性修复,并限制急性肾损伤向慢性肾脏病的转变。 研究者首先关注了PHD家族三个亚型(PHD1、PHD2、PHD3)在肾缺血后内皮细胞中的表达变化。人类单细胞测序数据显示,内皮细胞中PHD3显著上调,而PHD1和PHD2无明显变化。在另一个独立数据集中,PHD3同样显著上调。该发现在小鼠缺血再灌注损伤(IRI)模型中进一步得到证实:内皮PHD3蛋白于损伤后第2天开始升高,第14天达到峰值。 图1|人和小鼠肾损伤后的肾内皮PHD1、PHD2和PHD3的差异表达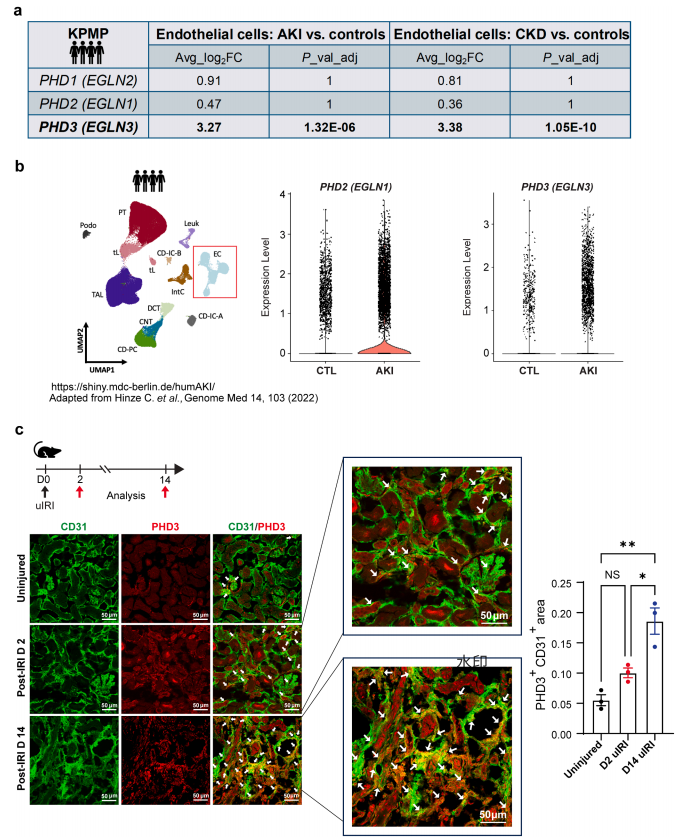
为明确PHD3在肾缺血后修复中的功能,研究者分别构建了内皮特异性敲除PHD1或PHD3的小鼠。结果显示,敲除PHD1对肾小管损伤、胶原沉积、促纤维化基因表达及毛细血管密度均无显著影响;而敲除PHD3则导致肾小管损伤加重、胶原沉积增加约42%、促纤维化基因表达升高,同时毛细血管密度减少约53%。这表明PHD3在肾缺血后修复中发挥着关键的保护功能。 图2 | 缺血后的内皮PHD1失活对肾纤维化无影响 图3 | 缺血后的内皮PHD3失活导致肾脏修复不良,伴随纤维化增加和毛细血管减少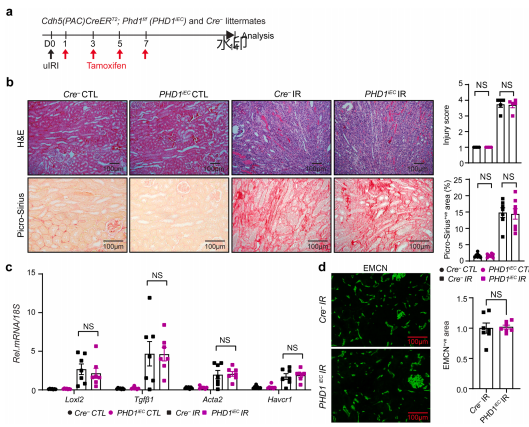
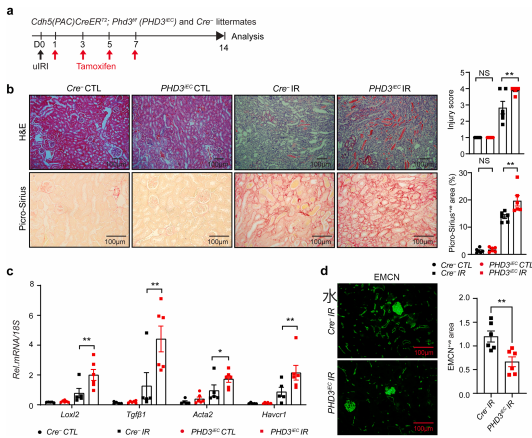
进一步研究发现,内皮PHD3失活会显著改变肾缺血后的炎症反应。与对照组相比,PHD3EC小鼠肾脏中促炎基因(Icam1、Vcam1、Cxcl10)及巨噬细胞标志物F4/80的mRNA水平显著升高,ICAM1和VCAM1蛋白分别增加2.5倍和3.5倍,巨噬细胞数量增加约50%。 图4|缺血后内皮PHD3失活诱导内皮细胞源性促炎反应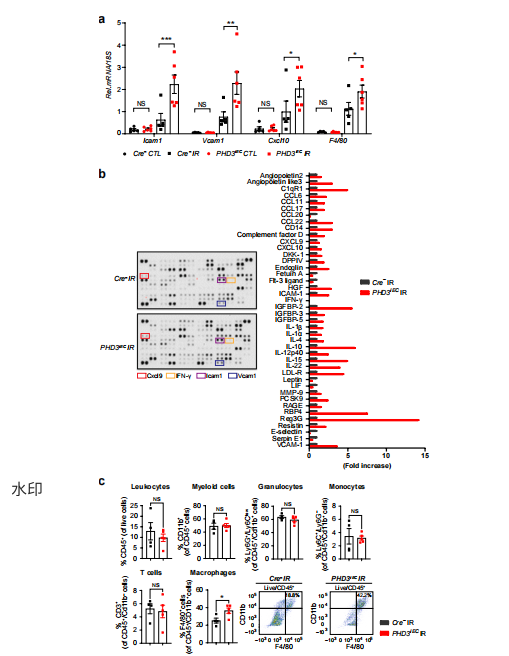
为从单细胞层面揭示内皮PHD3失活引发的内皮细胞特异性变化,研究者对PHD3杂合缺失小鼠缺血后的肾脏进行了单细胞RNA测序。结果显示,内皮细胞中有278个基因发生显著变化,上调最显著的通路包括干扰素γ应答、Myc靶基因及干扰素α应答,其中IFN-γ相关基因(如Ifitm3、Ifit3、Irf7)在内皮细胞中显著高表达。 图5 | 单细胞测序显示,内皮PHD3失活导致缺血后第14天肾脏内皮细胞出现干扰素-γ反应激活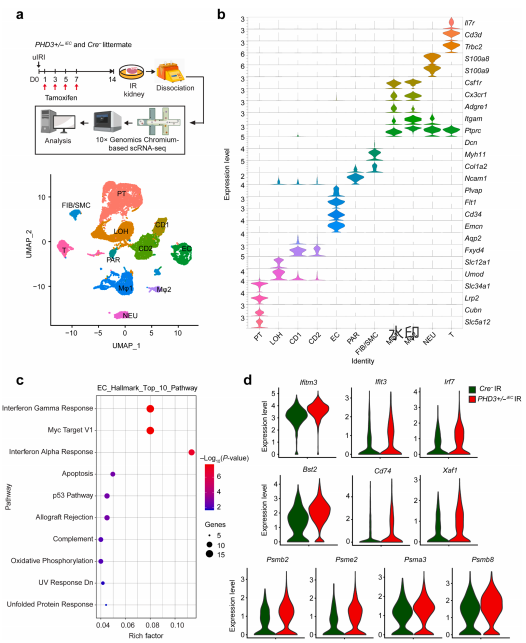
为验证PHD3调控炎症的机制,研究者进行了体外实验。在人肺动脉内皮细胞中敲低PHD3后,促炎基因表达显著升高;而同时敲低PHD3和ARNT后,这一效应被消除。相反,过表达PHD3则抑制上述基因的表达。这表明PHD3通过ARNT/HIF依赖的方式负向调控IFN-γ诱导的内皮促炎反应。 图6 | PHD3通过ARNT依赖的方式调控IFN-γ诱导的促炎基因转录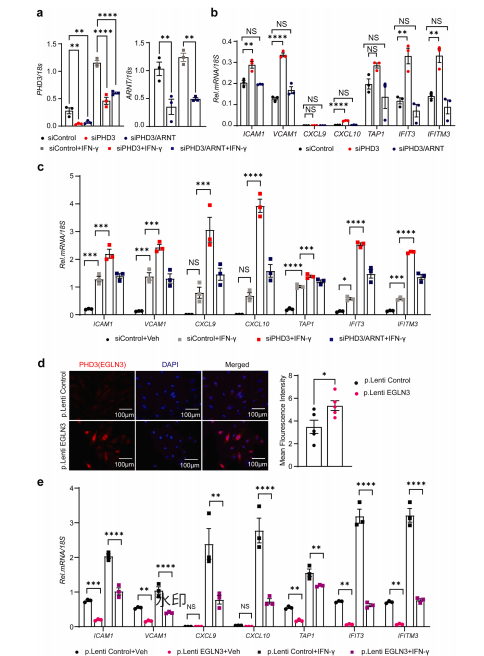
为在体内进一步验证HIF信号通路的必要性,研究者构建了内皮特异性双敲除PHD3和ARNT的小鼠。结果显示,与单敲PHD3不同,双敲除小鼠的肾小管损伤程度、胶原沉积面积、促纤维化基因(Loxl2、Tgfβ1、Havcr1)以及促炎基因(Vcam1、Icam1、Cxcl10)的表达均恢复至对照组水平。这表明同时敲除ARNT可挽救PHD3缺失导致的纤维化和炎症表型。 图7 | 缺血后内皮ARNT失活可阻断PHD3缺失诱导的促纤维化效应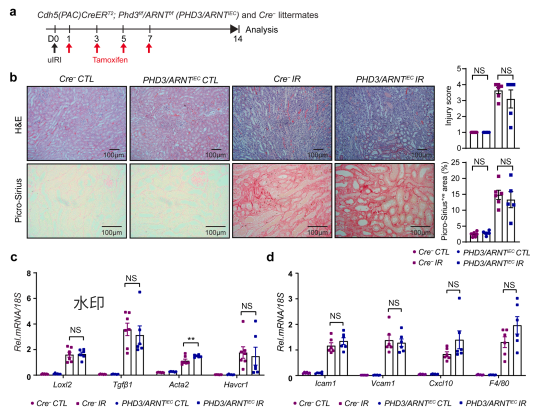
基于以上发现,本研究提出了一个整合性工作模型:肾脏缺血后,内皮细胞上调PHD3,通过羟基化作用限制HIF-α的稳定性,以HIF/ARNT依赖的方式对干扰素-γ信号通路施加“分子刹车”。这一机制可抑制内皮细胞的过度促炎活化及巨噬细胞招募,从而减轻炎症反应、保护微血管结构、限制纤维化进程,最终引导肾脏完成适应性修复,阻止其向慢性化方向演变。 图8|内皮PHD3依赖性调节缺血后肾脏修复的模型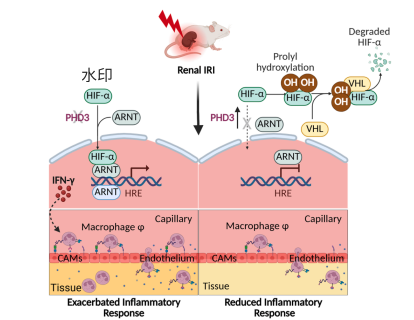
综上所述,本研究系统阐明了内皮细胞中PHD3通过HIF/ARNT轴抑制干扰素-γ信号,进而促进缺血后肾脏修复的完整机制。该研究不仅深化了对肾脏损伤修复机制的理解,也为开发靶向缺氧信号通路的新型肾脏病治疗方法提供了关键的理论基础和潜在的干预新靶点。
参考文献: Sharma R, Tiwari R, Zhou Y, O'Sullivan J, Maekawa H, Borkowski GS, Bukaric H, An SY, Thorp EB, Chandel NS, Quaggin SE, Kapitsinou PP. Endothelial prolyl hydroxylase 3 mitigates maladaptive inflammation to promote post-ischemic kidney repair. Kidney Int. 2026 Jun;109(6):1173-1187.